AnnData Concepts¶
QPX uses AnnData (.h5ad) as the primary format for expression views -- both Absolute Expression and Differential Expression. This page describes the general AnnData conventions shared across expression views.
Why AnnData?¶
AnnData is the standard interchange format for the scverse ecosystem (scanpy, scvi-tools, muon). It provides:
- Matrix-form representation (samples x proteins) natural for expression data
- Multi-layer storage for alternative quantifications (raw, normalized, log-transformed)
- Structured metadata for samples (
obs), proteins (var), and unstructured data (uns) - HDF5 backend for efficient storage and partial reads
- Ecosystem interop with scanpy plotting, scvi-tools, muon multi-omics, and Lamin.ai
AnnData structure overview¶
AnnData
X (n_obs x n_vars) Primary data matrix
obs (n_obs x ...) Observation (sample) metadata
var (n_vars x ...) Variable (protein) metadata
layers (n_obs x n_vars) Alternative quantification matrices
uns (dict) Unstructured metadata (DE results, file metadata)
obsm/varm (optional) Embeddings and dimensionality reductions
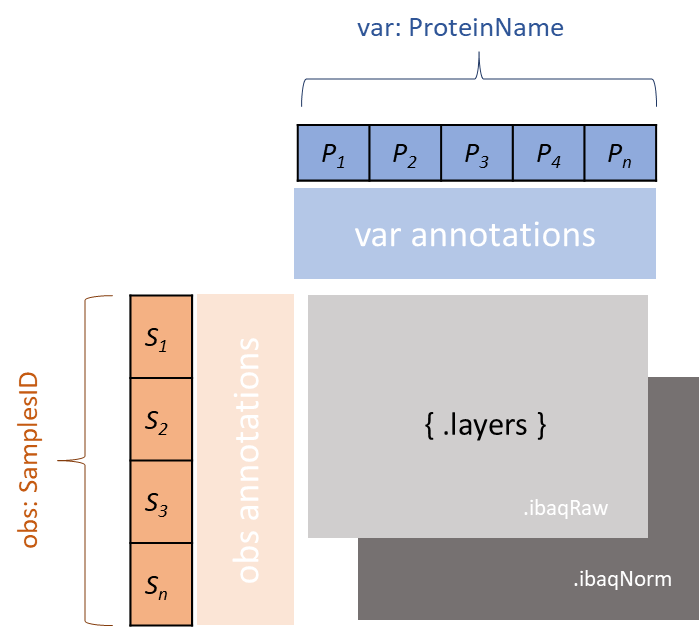
Conventions¶
obs index = sample accession¶
The obs index uses sample accessions from the SDRF. Sample-level metadata (organism, tissue, disease, replicate info) is stored as columns in obs.
var index = protein accession¶
The var index uses UniProt protein accessions. Protein-level metadata (gene names) is stored as columns in var.
X = primary quantification¶
The primary quantification matrix occupies X. For absolute expression, this is ibaq_log. The choice of log-transformed values as the primary matrix follows the scRNA-seq convention where X holds log-normalized counts.
layers = alternative quantifications¶
Additional quantification types are stored as AnnData layers. Each layer has the same dimensions as X (samples x proteins).
uns = file metadata + DE results¶
File-level metadata (QPX version, project accession, creation date) and differential expression results are stored in uns.
File naming¶
Expression AnnData files follow the QPX naming convention:
| View | Extension | Example |
|---|---|---|
| Absolute Expression | .ae.h5ad |
PXD000000.ae.h5ad |
| Differential Expression | .de.h5ad |
PXD000000.de.h5ad |
Reading and writing¶
Reading¶
import anndata as ad
# Read AE data
adata = ad.read_h5ad("PXD000000.ae.h5ad")
print(adata)
# AnnData object with n_obs x n_vars = 120 x 5432
# obs: 'organism', 'organism_part', 'disease', ...
# var: 'gene_name'
# layers: 'ibaq_raw', 'ibaq_ppb', 'copies_per_cell', 'concentration_nm'
Writing¶
Using the QPX library API (proposed)¶
# AE
ae = project.ae()
adata = ae.to_anndata(x_column="ibaq_log")
# DE
de = project.de()
adata.uns["de_results"] = de.to_de_results()
# Save combined
adata.write("PXD000000.ae.h5ad")
Missing values¶
When a protein is not detected in a sample, the corresponding cell in X and layers contains NaN. This is consistent with how scRNA-seq handles dropout events and is the standard behavior when pivoting from long-form to matrix-form.
Multi-omics integration¶
AnnData enables integration of proteomics data with other omics modalities:
- CITE-seq + bulk proteomics: Concatenate protein-level AnnData objects from different technologies.
- muon: Use
muon.MuDatato combine proteomics and transcriptomics AnnData objects. - Lamin.ai: Register QPX AnnData output as a Lamin Artifact with schema validation and ontology-backed labels.
Further reading¶
- Absolute Expression -- AE-specific AnnData schema and examples
- Differential Expression -- DE-specific AnnData schema and examples
- AnnData documentation -- Official AnnData reference
- scanpy -- Analysis framework for AnnData